
Синдром Ли — это редкое генетическое заболевание, которое проявляется множественными неврологическими и метаболическими нарушениями, что делает его диагностику и лечение сложными задачами для врачей и пациентов. В данной статье мы рассмотрим основные симптомы синдрома Ли, его типы наследования и методы диагностики, а также предложим рекомендации по эффективному лечению и профилактике. Понимание особенностей этого синдрома и доступных терапевтических подходов поможет пациентам и их семьям лучше справляться с заболеванием, а также повысит осведомленность медицинских специалистов о важности ранней диагностики и индивидуализированного подхода к лечению.
Классификация
Распределение различных форм синдрома Ли (болезнь Лея) зависит от расположения дефектного гена на хромосомах и нарушений в белках митохондриальной системы. Заболевание может быть связано с изменениями в Х-хромосомах, аутосомах и митохондриальной ДНК, охватывающей участки от первого до пятого комплекса (I, II, III, IV, V). Основные виды поражений, вызванные мутациями, включают:
- I комплекс (НАДН-KoQ) связан с изменениями в ядерных генах: NDUFA10, NDUFS4, NDUFS3, DUFAF2. Дефекты в митохондриальной ДНК, такие как MTND1,2,3, приводят к снижению синтеза аденозинтрифосфата (АТФ), который играет ключевую роль в клеточном энергетическом обмене. Наследуется двумя способами.
- II комплекс (сукцинат-KoQ) характеризуется мутациями на 5-й хромосоме в гене SDHA, что приводит к нарушению работы белков. Этот ген контролирует активность субъединицы А сукцинат-KoQ-редуктазы и участвует в метаболизме трикарбоновых кислот и дыхательной цепи. Передается только по аутосомно-рецессивному типу.
- III комплекс (KoQН2-цитохром с-редуктаза) связан с мутациями в гене BCS1L на 2-й хромосоме, что снижает способность гена преобразовывать наследственную информацию в белок. Заболевание возникает из-за дефектов убихинон-с-редуктазы. Наследуется аутосомно-рецессивным способом.
- IV комплекс (цитохром с-оксидаза) затрагивает мутации ядерных генов на 17-й хромосоме (SCO1, COS10) и нарушения в митохондриальной ДНК. Механизмы передачи до конца не изучены.
- V комплекс (АТФ-синтаза) характеризуется дефектом ATPAF2 на 17-й хромосоме, что приводит к снижению окислительных процессов в производстве аденозинтрифосфата.
Классификация включает болезнь Лея, вызванную редкой мутацией PDHA1, находящейся на Х-хромосоме. Эта аномалия затрагивает только мальчиков, а матери выступают носителями. Девочки могут унаследовать дефектный ген, но сами не заболеют; заболевание может проявиться у их сыновей.
Передача синдрома Ли по митохондриальному типу наследования касается исключительно женщин, которые являются носителями дефектных митохондрий. Каждая митохондрия содержит свою ДНК, и при делении яйцеклетки сложно определить распределение «больных» генов. Женщина, выглядящая здоровой, может представлять опасность для своего потомства.
Вероятность развития болезни зависит от количества неполноценных митохондрий, которые ребенок унаследует. Синдром может проявляться как у девочек, так и у мальчиков. Если мальчик не получил достаточное количество мутированных генов, он не представляет угрозы для своего потомства.
Врачи подчеркивают важность комплексного подхода к лечению синдрома Ли, редкого генетического заболевания, связанного с нарушениями обмена веществ. Основное внимание уделяется поддерживающей терапии, направленной на улучшение качества жизни пациентов. Специалисты рекомендуют регулярные консультации с неврологами и генетиками для мониторинга состояния и коррекции лечения. Физиотерапия и реабилитационные мероприятия играют ключевую роль в поддержании двигательной активности и снижении симптомов. Врачи также акцентируют внимание на необходимости индивидуального подхода, так как проявления синдрома могут значительно варьироваться. Важно учитывать психологические аспекты, обеспечивая поддержку как пациентам, так и их семьям.

Основные симптомы
К группе риска подострой некротизирующей энцефаломиелопатии относятся младенцы в возрасте до одного года, хотя в редких случаях заболевание может проявляться и в подростковом возрасте. Начальная клиническая картина синдрома Ли включает в себя:
- вялость и сонливость;
- возможную повышенную возбудимость;
- отказ грудных детей от кормления (отсутствие глотательного и сосательного рефлексов);
- недостаточный набор веса, соответствующего возрасту.
По мере прогрессирования заболевания наблюдаются следующие симптомы:
- непроизвольные движения (миоклония);
- задержка в моторном и психическом развитии;
- мышечная слабость, сменяющаяся гипертонусом;
- проявления пирамидной недостаточности;
- нарушения кислотно-щелочного баланса (ацидоз);
- увеличение размеров сердца и поражение миокарда;
- тремор;
- недостаточная вентиляция легких и поверхностное дыхание;
- снижение сухожильных рефлексов;
- потеря координации движений.
Ключевой особенностью синдрома Ли является его прогрессирующее течение. Эта форма заболевания характеризуется ярко выраженной симптоматикой:
- тонические судороги, охватывающие все тело;
- частичная или полная атаксия;
- нарушения зрительного нерва;
- парезы и потеря мышечной чувствительности;
- эпилептические приступы;
- утрата ранее приобретенных навыков;
- быстрое развитие слабоумия.
Недостаток аденозинтрифосфата затрагивает не только центральную нервную систему, но и внутренние органы. У детей заболевание может привести к увеличению сердца и печени, а также к развитию желтухи. В процессе прогрессирования болезни наблюдаются нарушения дыхательной функции, проявляющиеся симптомами Чейна-Стока (постепенное учащение дыхания), что, в свою очередь, вызывает гипервентиляцию легких и спазмы скелетной мускулатуры (тонические и клонические судороги). Обычно летальный исход наступает через 2–4 года из-за сердечной или дыхательной недостаточности.
 Читайте также:
Читайте также:

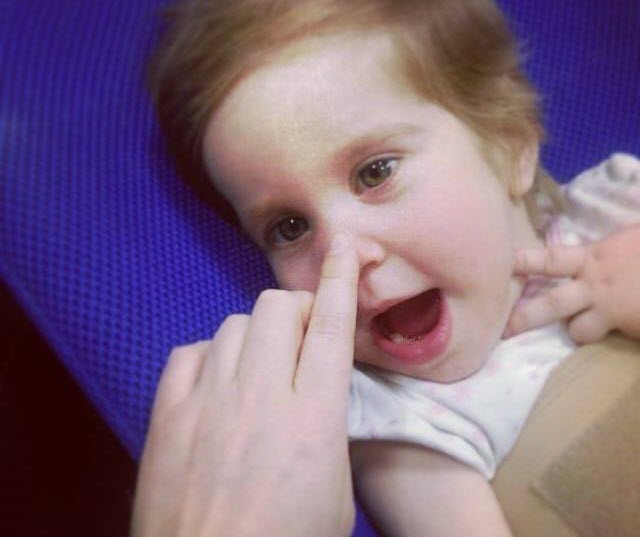

| Категория лечения | Метод/Препарат | Цель |
|---|---|---|
| Симптоматическая терапия | Коэнзим Q10 (убихинон) | Улучшение функции митохондрий, снижение окислительного стресса |
| Тиамин (витамин B1) | Поддержка метаболизма углеводов, особенно при дефиците | |
| Биотин (витамин B7) | Участие в метаболизме жирных кислот и аминокислот | |
| L-карнитин | Транспорт жирных кислот в митохондрии для производства энергии | |
| Дихлорацетат (DCA) | Снижение уровня лактата, улучшение функции митохондрий (применяется с осторожностью) | |
| Противосудорожные препараты | Контроль судорог (например, вальпроат, леветирацетам) | |
| Препараты для снижения спастичности | Уменьшение мышечного тонуса (например, баклофен) | |
| Лечение дистонии | Ботулинический токсин, антихолинергические препараты | |
| Поддерживающая терапия | Физиотерапия, эрготерапия, логопедия | |
| Диетическая терапия | Кетогенная диета | Изменение метаболизма, использование кетоновых тел в качестве источника энергии |
| Диета с ограничением углеводов | Снижение нагрузки на митохондриальные пути метаболизма углеводов | |
| Добавки с жирными кислотами | Обеспечение источника энергии, поддержка клеточных мембран | |
| Экспериментальные/Исследовательские подходы | Генная терапия | Коррекция генетических дефектов, вызывающих синдром Ли (на стадии исследований) |
| Трансплантация митохондрий | Замена поврежденных митохондрий здоровыми (на стадии исследований) | |
| Фармакологические шапероны | Стабилизация белков, участвующих в функции митохондрий (на стадии исследований) | |
| Антиоксиданты | Снижение окислительного стресса (например, витамин Е, аскорбиновая кислота) | |
| Общие рекомендации | Регулярное наблюдение у специалистов | Невролог, генетик, метаболический специалист |
| Избегание стрессовых факторов | Инфекции, голодание, переохлаждение, которые могут усугубить состояние | |
| Поддерживающая терапия | Психологическая поддержка для пациента и семьи |
Причины патологии
Этиология прогрессирующего заболевания центральной нервной системы связана с мутациями в генах, которые отвечают за синтез белков, участвующих в энергетическом обмене различных комплексов, включая дыхательную цепь митохондрий, где происходит окислительное производство АТФ. В клетках живых организмов присутствует множество древних симбионтов с автономной ДНК, основная задача которых — выработка жизненно важной энергии.
Процесс окислительного фосфорилирования осуществляется в комплексах дыхательной цепи митохондрий, после чего электроны никотинамидадениндинуклеотида (NADH) переносятся органеллами в межмембранное пространство. Нейроглия, чувствительная к нехватке энергии, может инициировать аномальные процессы в центральной нервной системе, что приводит к различным нарушениям, проявляющимся в раннем детском возрасте.
В дополнение к генетическим мутациям, недостаток фермента может быть вызван рядом факторов:
- заболевания центральной нервной системы;
- наличие опухоли или кисты в мозжечке;
- образование глиальных клеток (заменителей поврежденных нейронов) в головном или спинном мозге;
- чрезмерное образование новых сосудов, превышающих объем тканей (пролиферация);
- гибель нейронов в результате воспалительных процессов;
- наследственная предрасположенность (генетическая детерминанта).
Развитию патологии может способствовать любая инфекция или неправильно проведенная вакцинация.
Синдром Ли — редкое генетическое заболевание, которое вызывает множество вопросов и тревог у пациентов и их семей. Многие люди, столкнувшиеся с этой проблемой, отмечают, что рекомендации по лечению часто бывают недостаточно ясными и разнообразными. Некоторые врачи предлагают комплексный подход, включающий физиотерапию и поддерживающую терапию, что помогает улучшить качество жизни. Однако пациенты также делятся опытом о том, как важна индивидуализация лечения, так как симптомы могут сильно варьироваться. Важным аспектом является поддержка со стороны сообщества, где люди обмениваются информацией о новых методах и исследованиях. Многие находят утешение в том, что существуют специализированные клиники и группы поддержки, которые помогают справиться с эмоциональными и физическими трудностями, связанными с синдромом.
Диагностические исследования
Определение синдрома Ли осуществляется только в условиях стационара, так как амбулаторные методы не подходят. На начальном этапе важно провести дифференциацию этого заболевания от других патологий, которые могут иметь схожие клинические проявления:
- энцефаломиелит;
- амавротическая идиотия;
- заболевания инфекционно-аллергического характера;
- миоклонус-эпилепсия;
- лейкодистрофии.
Во время визуального осмотра особое внимание уделяется неврологическим симптомам:
- тремор конечностей;
- задержка в психомоторном развитии;
- недостаточная масса тела;
- фасцикуляция языка.
Диагностика включает в себя тщательное изучение наследственного фактора, особенно если заболевание имеет аутосомно-рецессивный тип наследования. В случае мутации генов невозможно установить, передалась ли патология от матери к ребенку. В таких ситуациях рекомендуется провести молекулярный анализ.
Далее проводятся исследования с использованием специализированного оборудования:

- С помощью магнитно-резонансной томографии в режимах FLAIR и T2 выявляются поражения продолговатого мозга, таламуса и базальных ганглиев, которые особенно чувствительны к гипоксии. Патологические изменения имеют симметричный характер. В некоторых случаях могут наблюдаться аномалии в белом веществе полушарий, вызванные кистозными образованиями.
- Молекулярная диагностика используется для определения типа мутации генов SURF1 и BCS1L.
- Электрокардиография.
- Офтальмоскопия для исследования глазного дна.
- Электронейромиография позволяет выявить разрушение миелина в белом веществе периферической нервной системы (демиелинизацию) из-за снижения скорости передачи нервных импульсов. На ЭКГ фиксируется преобладание медленной активности волн.
Также производится забор спинномозговой жидкости для определения уровня белка и лимфоцитарного плеоцитоза. Биохимическое исследование крови показывает высокий уровень кетоновых тел, что указывает на наличие лактат-ацидоза.
Эффективные методы лечения
Специальные методы лечения некротизирующей энцефалопатии пока не разработаны. Основные усилия направлены на улучшение функционирования центральной нервной системы и имеют симптоматический характер, чтобы стабилизировать состояние ребенка. В процессе терапии используются следующие группы медикаментов:
- Антибактериальные препараты – «Цефтриаксон»;
- Противогрибковые средства – «Флюконазол», «Дифлюкан»;
- Противовирусные лекарства – «Завиракс»;
- Антибиотики – «Биотин», «Ампициллин»;
- Витамины – С, В1, Е 200, В6 («Пиридоксин»);
- Средства для нормализации состояния – «Тиамин-хлорид», «Кокарбоксилаза»;
- Глюкокортикостероиды – «Дексаметазон», «Преднизолон»;
- Нейротропные препараты – «Нейромидин», «Пирацетам»;
- Препараты для снятия судорог – «Клобазам», «Эпилим», «Клоназепам»;
- Гормональные средства – «Гидрокортизон», «Тетракозактид»;
- Метаболические препараты – «Тиамин», «L-карнитин», «Рибофлавин», «Коэнзим Q10», «Актовегин», «Церебролизин»;
- Диуретики – «Фуросемид»;
- Кардиометаболические средства – «Кардио – АБВ», «Милдронат»;
- Муколитики – «Амброксол»;
- Препараты для нормализации кишечной микрофлоры – «Креон», «Линекс»;
- Средства на основе вальпроевой кислоты.
При медикаментозном лечении данной патологии важно соблюдать диету, которая предполагает ограничение продуктов с высоким содержанием белка, с нормой поступления не более 1 грамма в сутки. Если в семье есть ребенок с болезнью Лея, настоятельно рекомендуется проконсультироваться с врачом для получения первой помощи в случае приступа.

Прогноз и рекомендации по профилактике
После зачатия, если имеются основания полагать о генетическом наследии, важно провести перинатальную диагностику плода. Цель данного исследования заключается в выявлении анеуплоидий, таких как синдромы Дауна, Ли и Шерешевского-Тернера. В случае положительного результата беременность может быть прервана. Дети с болезнью Лея, как правило, живут от 2 до 5 лет. Симптоматическая терапия может замедлить развитие заболевания, но не устраняет его. При хронической форме нарушения это может привести к инвалидности и летальному исходу из-за сердечной и дыхательной недостаточности. Профилактика данной патологии включает в себя медико-генетическую консультацию обоих родителей перед началом беременности.
Поддерживающая терапия и реабилитация
играют ключевую роль в управлении синдромом Ли, который является редким наследственным заболеванием, вызываемым мутациями в генах, отвечающих за метаболизм митохондрий. Основная цель поддерживающей терапии заключается в улучшении качества жизни пациентов и замедлении прогрессирования заболевания.
Одним из важнейших аспектов поддерживающей терапии является обеспечение адекватного питания. Пациенты с синдромом Ли часто страдают от недостатка энергии и могут иметь проблемы с усвоением питательных веществ. Рекомендуется консультация с диетологом, который поможет разработать индивидуальный план питания, включающий высококалорийные и питательные продукты, а также добавки, содержащие витамины и минералы, необходимые для поддержания метаболических процессов.
Физическая реабилитация также является важной частью поддерживающей терапии. Регулярные физические упражнения могут помочь улучшить мышечный тонус, координацию и общую физическую выносливость. Рекомендуется разработать программу упражнений, адаптированную к возможностям пациента, включая легкие аэробные нагрузки, растяжку и силовые тренировки. Физиотерапевты могут помочь в создании безопасной и эффективной программы реабилитации.
Психологическая поддержка не менее важна. Пациенты с синдромом Ли и их семьи могут столкнуться с эмоциональными и психологическими трудностями, связанными с заболеванием. Психотерапия, группы поддержки и консультации могут помочь справиться с тревогой, депрессией и другими эмоциональными проблемами. Важно создать поддерживающую среду, где пациенты могут открыто обсуждать свои чувства и переживания.
-
Читайте также:

Лекарственная терапия может быть назначена для управления симптомами, такими как судороги, боли и другие сопутствующие состояния. Важно, чтобы лечение проводилось под контролем опытного врача, который сможет адаптировать терапию в зависимости от индивидуальных потребностей пациента.
Наконец, регулярное наблюдение у специалистов, таких как неврологи и генетики, необходимо для мониторинга состояния пациента и корректировки лечения по мере необходимости. Это позволит своевременно выявлять изменения в состоянии здоровья и адаптировать терапию, что может существенно повлиять на качество жизни пациента.
Таким образом, поддерживающая терапия и реабилитация при синдроме Ли требуют комплексного подхода, включающего медицинские, физические и психологические аспекты. Индивидуализированный план лечения, основанный на потребностях пациента, может значительно улучшить его состояние и качество жизни.
Вопрос-ответ
Можно ли жить с синдромом Ли?
Многие пациенты с синдромом Ли могут умереть от дыхательной недостаточности в детстве, но те, у кого заболевание протекает в менее тяжелой форме и начинается позже, могут дожить до подросткового возраста или начала двадцатых годов. К сожалению, лекарства от синдрома Ли не существует.
Синдром ли прогноз?
Каковы шансы на выздоровление от болезни Лея? Прогноз для людей с болезнью Лея плохой. Люди, у которых отсутствует активность митохондриального комплекса IV, и люди с дефицитом пируватдегидрогеназы, как правило, имеют худший прогноз и умирают в течение нескольких лет.
Советы
СОВЕТ №1
Обратитесь к специалисту. При первых симптомах синдрома Ли важно проконсультироваться с неврологом или генетиком, чтобы получить точный диагноз и рекомендации по лечению.
СОВЕТ №2
Следите за питанием. Правильное питание может помочь поддерживать уровень энергии и общее состояние здоровья. Обсудите с врачом, какие продукты лучше включить в рацион.
СОВЕТ №3
Регулярные физические упражнения. Легкие физические нагрузки могут улучшить мышечный тонус и общее самочувствие. Проконсультируйтесь с физиотерапевтом для разработки индивидуальной программы упражнений.
СОВЕТ №4
Поддерживайте эмоциональное здоровье. Психологическая поддержка и участие в группах поддержки могут помочь справиться с эмоциональными трудностями, связанными с синдромом Ли.




